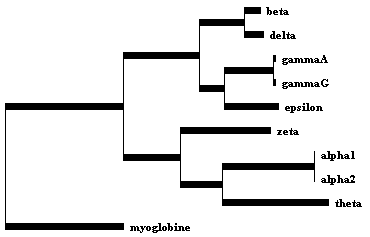
Les différentes hémoglobines humaines sont constituées de plusieurs chaînes dont les gènes sont situés sur 2 chromosomes :
- Sur le chromosome 11, on trouve les gènes des chaînes beta, gamma A, gamma G, delta et epsilon.
- Sur le chromosome 16, on trouve les gènes des chaînes alpha 1, alpha 2, zeta et theta.
- On peut y ajouter le gène de la myoglobine qui se trouve sur le chromosome 22.
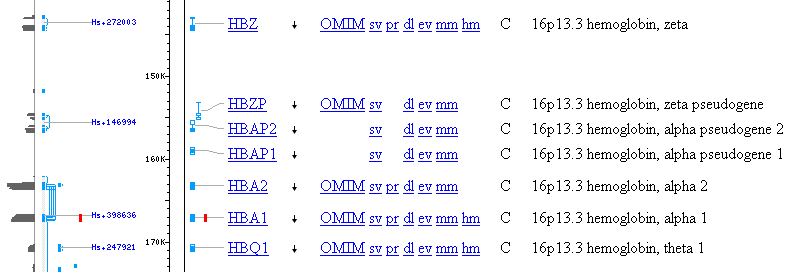
Chromosome 11 :

Chromosome 16 :
Les hémoglobines adultes sont HbA (plus de 95%) formée de 2 chaînes alpha et de 2 chaînes beta et HbA2 formée de 2 chaînes alpha et de 2 chaînes delta.
L’hémoglobine fœtale HbF est formée de 2 chaînes alpha et de 2 chaînes gamma.
Les hémoglobines embryonnaires sont Gower 1 formée de 2 chaînes zeta et de 2 chaînes epsilon, Gower 2 formée de 2 chaînes alpha et de 2 chaînes epsilon, Portland formée de 2 chaînes delta et de 2 chaînes gamma.
Les molécules étudiées ici proviennent du site NCBI (National Center for Biotechnology Information).
Traitement des données dans Anagène
Ces molécules ont été regroupées dans un fichier qui s’ouvre dans Anagène, globines.edi.
Le fichier est zippé, il faut l’extraire pour pouvoir l’utiliser.
L’ordre qui a été choisi est l’ordre décroissant de longueur des molécules ce qui permet d’éviter des erreurs dans le dénombrement des différences.
Faire un alignement avec discontinuités de toutes ces molécules.
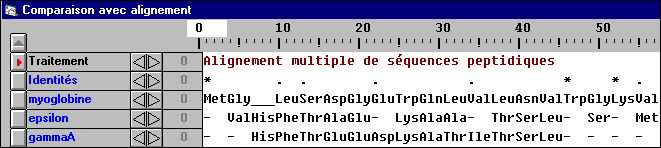
La ligne traitement est pointée par défaut, lire les informations sur la ligne pointée (touche F2).
On a alors le nombre d’acides aminés de la première molécule (au début, la myoglobine) puis, pour chaque molécule, le nombre d’acides aminés identiques à la molécule de référence ce qui, par soustraction, donnera le nombre de différences. Collecter le nombre de ces différences pour chaque molécule.
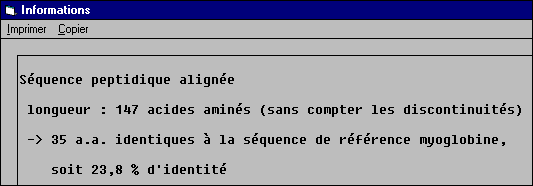
Les informations données par la ligne Identités montrent que, si le nombre d’acides aminés identiques ne représente qu’environ 12% lorsque toutes les molécules sont alignées, ce pourcentage passe à 23 lorsque l’on élimine la myoglobine, ce qui révèle une certaine proximité de la structure primaire de ces molécules.
Traitement des données dans Phylogène
Les séquences moléculaires ont été regroupées dans le fichier globines.pir à télécharger par un clic droit. Le fichier est zippé, il faut l’extraire pour pouvoir l’utiliser.
Un fichier .pir est au format texte, sa structure est donc visible dans un éditeur de texte, il contient les noms des molécules (après le signe >) et la séquence à la ligne au format une lettre.
Ce fichier s’ouvre dans Phylogène comme un tableau de molécules en prenant soin de choisir la catégorie de molécules non alignées.
Il suffit alors de faire aligner les molécules par le logiciel, on peut alors obtenir la matrice de distance et faire tracer l’arbre.
La matrice est un peu différente de celle obtenue "à la main" dans Anagène mais l’arbre n’est pas différent si l’on a choisi dans les options la méthode NJ (Neighbor Joining).