La cytométrie en flux, la PCR quantitative ou Q-PCR, la bioinformatique et l’annotation des génomes.
La cytométrie en flux (Fluorescence Activated Cell Sorting)
Présentation par Chloé Journo, Agrégée préparatrice à l’ENS
Propos rapportés par Georges Grousset
L’approche microscopique des caractéristiques cellulaires si on vise à les identifier cellule par cellule est longue, très lourde et délicate (possibilité d’erreur liée à l’observation). Le cytomètre de flux (FACS) est un outil d’analyse automatique rapide, puissant, sûr et permettant d’identifier cellule par cellule telle ou telle caractéristique. Il permet d’analyser des paramètres sur la cellule vivante et des perspectives immenses s’ouvrent lorsque les cellules sont marquées spécifiquement.
Le cytomètre comporte une chambre de lecture éclairée par un rayon laser devant lequel les cellules passent une à une.
Deux paramètres sont mesurables directement, sans marquage cellulaire :
– la taille de la cellule : plus la cellule est grosse, plus la diffusion du rayon, dans son axe, est grande (diffusion axiale ou FSC).
– sa granularité : plus la cellule est granuleuse, plus la diffraction du rayon observée latéralement est grande (diffusion latérale ou SSC).
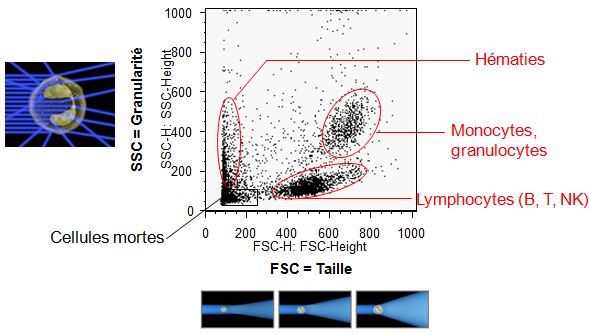
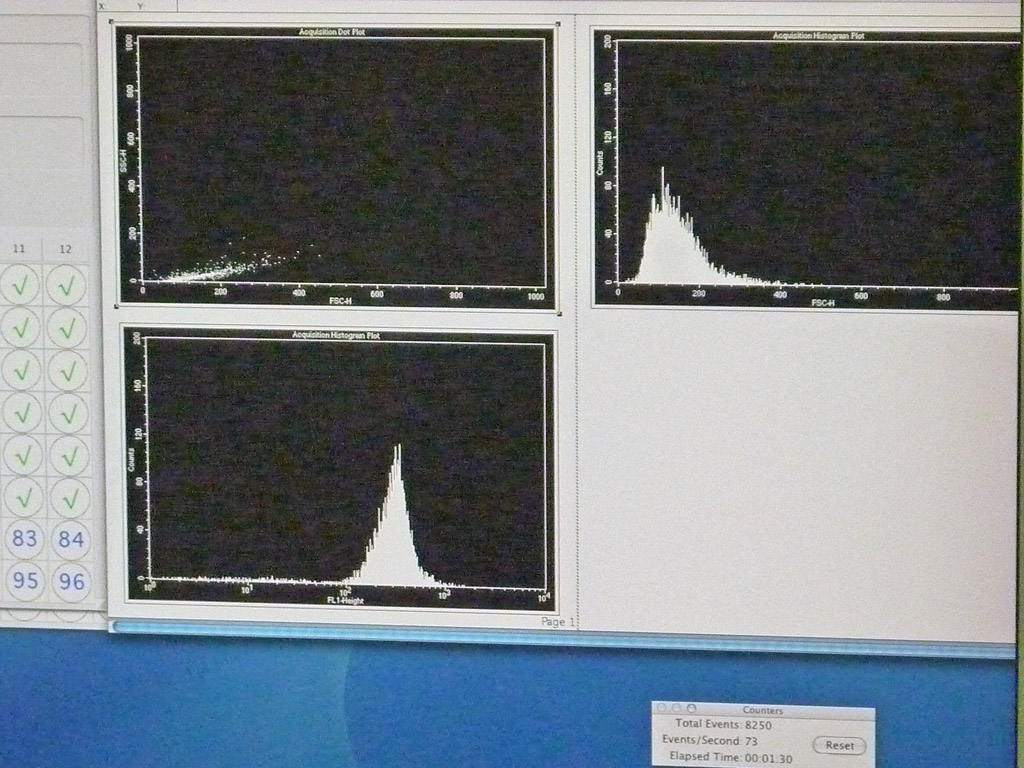
L’afficheur numérique des résultats produit un graphique avec SSC en ordonnées et FSC en abscisses. L’analyse d’un échantillon porte en général sur un nombre de cellules allant de 10000 à 100000. Chaque point sur le diagramme correspond aux caractéristiques d’une seule cellule.
|
Diagramme ci-contre obtenu avec un échantillon de sang
– FSC faible : petites cellules -> des hématies
– FSC grand et SSC faible : cellules plus grosses, peu granuleuses -> des lymphocytes
– FSC grand et SSC grand : grosses cellules, granuleuses -> des granulocytes
|
Le marquage spécifique par anticorps fluorescent permet de mesurer des paramètres indirects.
Le cytomètre prélève chaque échantillon dans des puits d’une cuve à échantillons, sur lequel un certain nombre de paramètres sont analysés puis passe à un second échantillon cellulaire ayant subi une préparation différente ou issu d’une population cellulaire particulière : il s’agit la du protocole expérimental décidé par le chercheur et non plus de la technique de cytométrie.
Ainsi, on peut fixer sur les cellules un marqueur particulier repéré par sa fluorescence : des diagrammes de fluorescence par l’utilisation de Fluoresceine Iso Thio Cyanate (FITC) sont obtenus avec le nombre de cellules ayant telle ou telle intensité de fluorescence (le diagramme peut être unimodal lorsque la population est homogène mais aussi multimodal, signe de l’existence de différents types cellulaires. L’analyse multiparamétrique (actuellement on peut détecter jusqu’à 19 paramètres différents et ainsi distinguer des populations de cellules les unes des autres) est donc possible en faisant varier la longueur d’onde d’excitation, grâce aux filtres séparant les longueurs d’ondes issues de l’échantillon soumis au rayon laser.
Les cellules repérées selon leurs caractéristiques, peuvent aussi être triées dans un champ électrique après avoir été chargées positivement ou négativement ou non chargées. Ainsi différentes populations de cellules peuvent être séparées.