Exploitation de ressources documentaires et analyse de séquences génétiques pour déterminer la cause d’une maladie musculaire chez un patient et réaliser une prédiction génétique.
Auteur : Vincent GUILI, Lycée Descartes, Saint-Genis-Laval
Liens avec le BO
BO spécial n°8 du 25 juillet 2019
Terminale spécialité : Corps humain et santé
Produire le mouvement : contraction musculaire et apport d’énergie
La cellule musculaire : une structure spécialisée permettant son propre raccourcissement
Connaissances
Dans certaines myopathies, la dégénérescence des cellules musculaires est due à un défaut dans les interactions entre les protéines membranaires des cellules et la matrice extra-cellulaire.
Capacités, attitudes
Remobiliser les acquis sur la matrice extracellulaire à travers l’exemple d’une myopathie.
Et remobilisation des acquis de première spécialité :
Liens avec le BO
BO spécial n°1 du 22 janvier 2019
Première : La Terre, la vie et l’organisation du vivant
Transmission, variation et expression du patrimoine génétique
Mutations de l’ADN et variabilité génétique
Les mutations sont à l’origine de la diversité des allèles au cours du temps. Selon leur nature elles ont des effets variés sur le phénotype.
L’expression du patrimoine génétique
Le code génétique est un système de correspondance, universel à l’ensemble du monde vivant, qui permet la traduction de l’ARN messager en protéines.
Le phénotype résulte de l’ensemble des produits de l’ADN (protéines et ARN) présents dans la cellule. Il dépend du patrimoine génétique et de son expression.
Capacités
Caractériser à l’aide d’un exemple les différentes échelles d’un phénotype (moléculaire, cellulaire, de l’organisme).
Mise en situation
Les dystrophies musculaires congénitales (DMC) constituent un ensemble de maladies caractérisées par une atteinte musculaire ("dystrophie") entrainant une faiblesse musculaire présente à la naissance ou apparaissant dans les premiers mois de la vie ("congénitale"). Toutes les DMC sont des maladies d’origine génétique : elles sont dues à des anomalies de l’ADN. Jusqu’à présent, près de 40 gènes (codant autant de protéines) différents impliqués dans les DMC ont été identifiés.
Une grande partie des protéines codées par ces gènes intervient dans les interactions entre la cellule musculaire et son environnement immédiat et préserve les cellules musculaires du stress mécanique lors des contractions musculaires.
Source : AFM Téléthon
Consigne
On dispose de l’arbre généalogique d’une famille touchée par une myopathie, ainsi que des analyses génétiques d’un des enfants atteints. D’autres documents présentent l’organisation moléculaire de la jonction entre la cellule musculaire et son environnement et la description de quatre myopathies d’origines différentes.
À partir de l’analyse de l’ensemble des documents, déterminer la myopathie qui touche cette famille et calculer le risque que l’enfant à naître soit atteint de myopathie.
Ressources
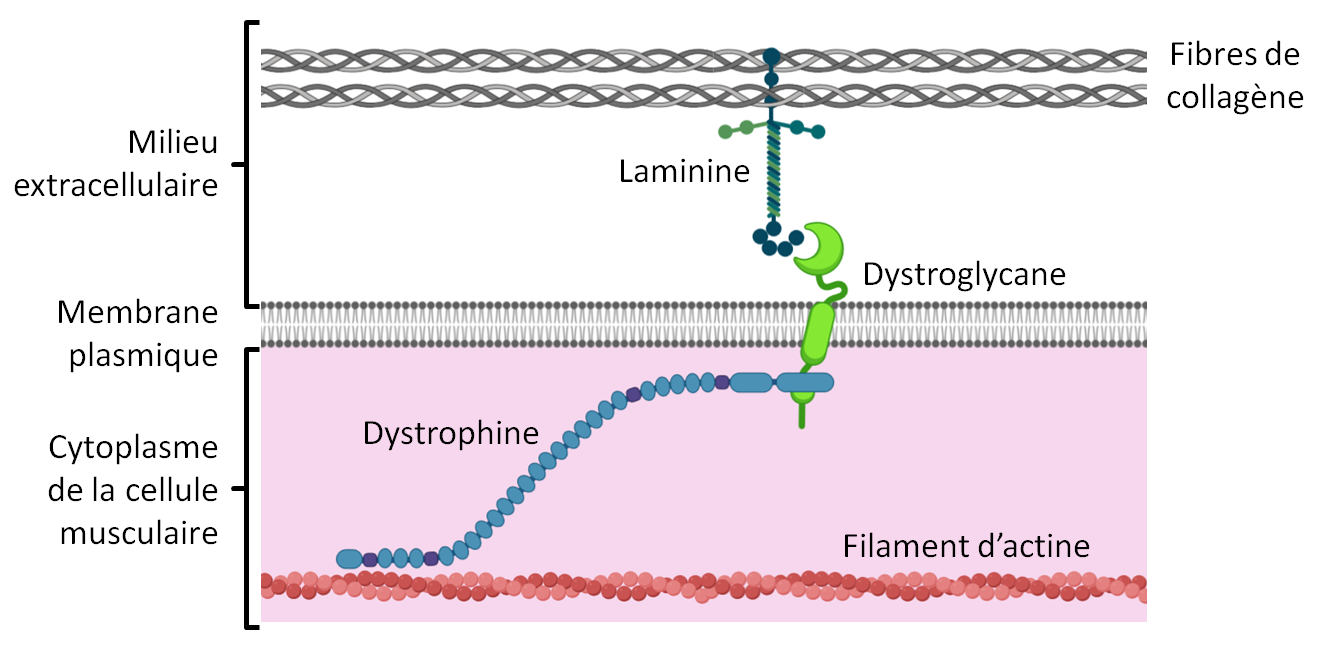
Le fonctionnement normal des muscles repose non seulement sur l’intégrité de la cellule musculaire et de ses filaments d’actine et de myosine, mais aussi sur la liaison de ces filaments avec les fibres qui entourent les cellules musculaires. Ces fibres, principalement constituées de collagène, forment la matrice extracellulaire. Les principales protéines qui relient l’actine au collagène sont la dystrophine, le dystroglycane et la laminine.
Document 1 : Organisation simplifiée des relations cellule musculaire - matrice extracellulaire
Échelles non respectées - document produit avec Biorender.
Myopathie d’Ullrich
La maladie est due à des mutations des gènes COL6A1, COL6A2, ou COL6A3 codant pour le collagène. Elle se caractérise par une faiblesse des muscles, des contractures (en particulier des coudes et des genoux), et une hyperextensibilité des articulations des mains, chevilles, pieds et doigts. Un retard de croissance et un déficit respiratoire sont fréquents. La probabilité d’être porteur d’un allèle muté est de 0,002.
Dystrophie musculaire associée à LAMA2
La maladie est due à des mutations du gène LAMA2 codant pour la laminine. Elle se caractérise par un faible tonus musculaire dès la naissance, le développement de contractures des grosses articulations, et une atteinte respiratoire progressive. L’atrophie musculaire et la faiblesse sévère empêchent généralement l’acquisition d’un déplacement autonome. La probabilité d’être porteur d’un allèle muté est de 0,002.
Syndrome de Walker-Warburg
La maladie est due à des mutations du gène DAG2 codant pour le dystroglycane. Elle se caractérise par un faible tonus musculaire, une faiblesse musculaire, un développement psychomoteur absent ou très pauvre, une atteinte oculaire et des convulsions. La probabilité d’être porteur d’un allèle muté est de 0,008.
Myopathie de Duchenne
La maladie est due à des mutations du gène DMD codant pour la dystrophine. Elle touche principalement les garçons. Elle se caractérise par une dégénérescence des muscles chez l’enfant. La faiblesse musculaire se propage à l’ensemble du corps jusqu’à une insuffisance respiratoire ou cardiaque fatale. La probabilité d’être porteur d’un allèle muté est de 0,03.
Document 2 : Principales maladies héréditaires touchant les muscles (myopathies)
Source : Orphanet
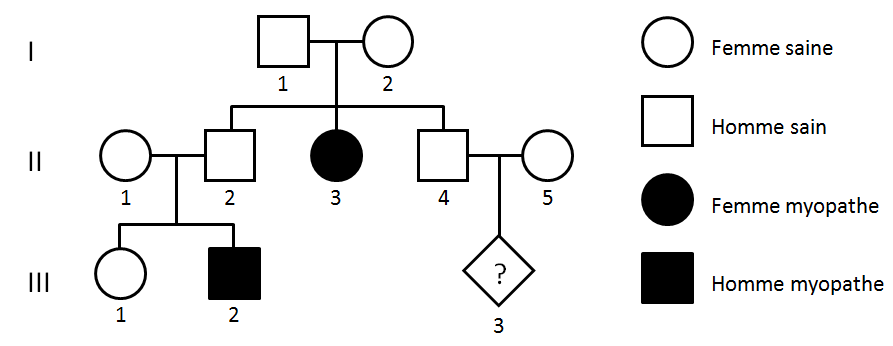
Document 3 : Arbre généalogique d’une famille présentant des enfants myopathes

Document 4 : Analyses génétiques sur l’enfant myopathe III.2
Fichier de séquence :

Myopathie.edi (clic droit "Enregistrer la cible du lien sous...")
ouverture directe dans Geniegen2 : https://www.pedagogie.ac-nice.fr/svt/productions/geniegen2/?load=EXT-MYOPATHIE
Résultats
1 - Traitement des séquences :
| Gène | Analyse des allèles | ||
| COL6A1 | Les allèles 1 et 2 sont identiques à l’allèle de référence => pas de mutation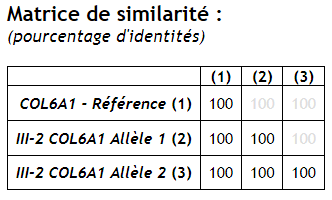
| ||
| LAMA2 | Les allèles 1 et 2 sont identiques à l’allèle de référence => pas de mutation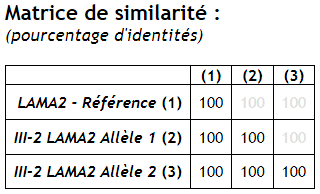
| ||
| DMD | Les allèles 1 et 2 sont différents de l’allèle de référence => 5784A->C Après traduction puis comparaison des protéines, on voit que la mutation ne crée aucune différence dans la protéine. GCA -> GCC en position 5784 ; ALA1928 -> ALA. C’est une mutation silencieuse due à la redondance du code génétique. 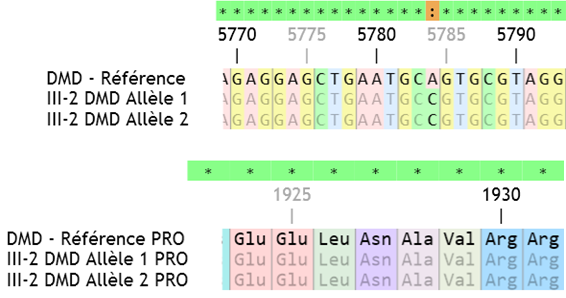
| ||
| DAG2 | Les allèles 1 et 2 sont différents de l’allèle de référence => 70C->T Après traduction puis comparaison des protéines, on voit que la mutation crée un codon STOP au début de la protéine : CAG->TAG ; 24GLN->STOP 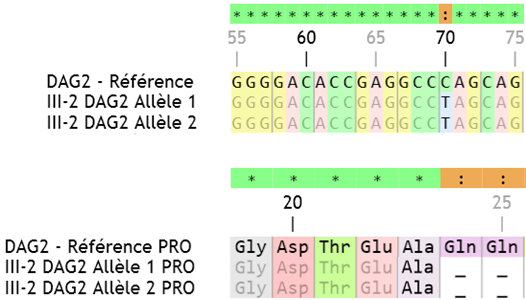
|
La myopathie qui touche cette famille est due à une mutation dans le gène DAG2 empêchant la production du dystroglycane : il s’agit donc du syndrome de Walker-Warburg.
2 - Calcul du risque :
Des individus sains ont des enfants malades, l’allèle responsable est donc récessif. Les individus III.2 et II.3, malades, sont homozygotes récessifs, leurs parents sont donc hétérozygotes. Donc I.1 et I.2 sont hétérozygotes.
Pour que l’enfant III.3 soit malade, il faut que ses parents II.4 et II.5 soient hétérozygotes.
On en déduit PIII.3 = 1/4 x PII.4 x PII.5
Les individus I.1 et I.2 sont hétérozygotes car ils ont eu un enfant atteint (II.3). On peut dès lors réaliser un tableau de croisement pour déterminer la probabilité de II.4 d’être hétérozygote :
| Gamètes parentaux | (DAG2/) | (dag2/) | |
| (DAG2/) | (DAG2//DAG2) | (DAG2//dag2) | |
(dag2/) |
(DAG2//dag2) | (dag2//dag2) |
II.4 est sain, donc la probabilité qu’il soit hétérozygote est 2/3 (66,6 %).
Pour II.5, en absence d’informations sur sa famille, on retient la probabilité générale dans la population, soit PII.5 = 0,008 (cf doc 2).
Donc PIII.3 = 1/4 x PII.4 x PII.5 = 1/4 x 2/3 x 0,008 = 0,00013 = 1/750
Références
- COL6A1 : séquence extraite de Genbank : https://www.ncbi.nlm.nih.gov/nuccore/NM_001848.3 - chromosome 21
- LAMA2 : séquence extraite de Genbank : https://www.ncbi.nlm.nih.gov/nuccore/NM_000426.4 - chromosome 6
- DAG2 : séquence extraite de Genbank : https://www.ncbi.nlm.nih.gov/nuccore/BC025702.1 - chromosome 17
- DMD - séquence de référence extraite du dossier ACCES - chromosome X

