Cette maladie est la plus fréquente des maladies héréditaires autosomiques récessives graves dans les populations d’origine européenne. Elle touche en moyenne un nouveau-né sur 2500 avec une fréquence variable selon l’origine géographique et ethnique des patients. En Europe, selon les régions, un enfant pour 1800 à 3500 naissances vivantes est atteint.
Avant la découverte de traitements, cette maladie était mortelle avant l’âge de la puberté et l’on peut se demander pourquoi, dès lors que ces individus ne procréaient pas, l’allèle est encore aussi fréquent dans l’espèce humaine.
Cette étude consiste à rechercher des arguments permettant de tenter d’expliquer cette dernière remarque.
La mutation
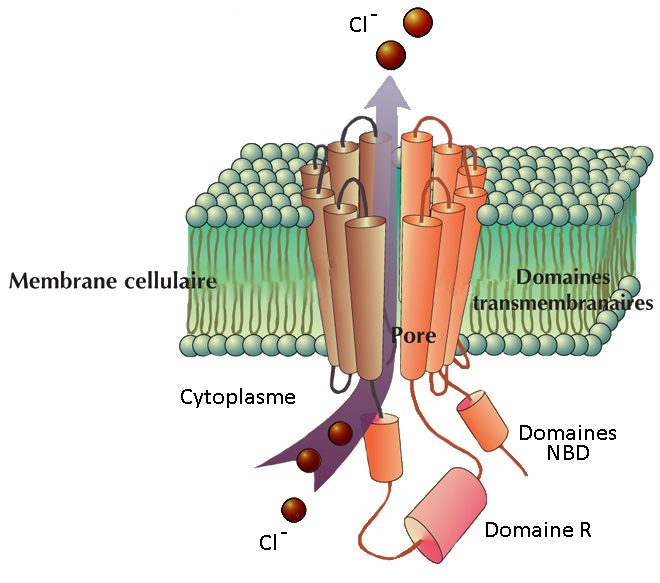
Le gène, porté par le chromosome 7 (7q31), a 4443 nucléotides et code pour une protéine de 1480 acides aminés dite CFTR (Cystic Fibrosis Transmembrane Regulator) qui forme un canal chlore.
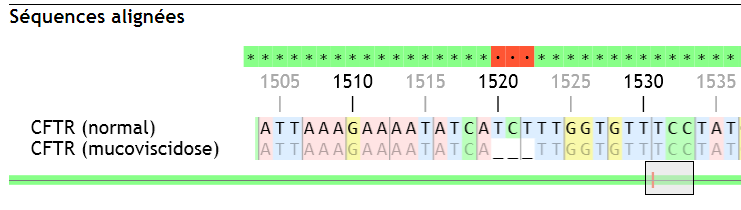
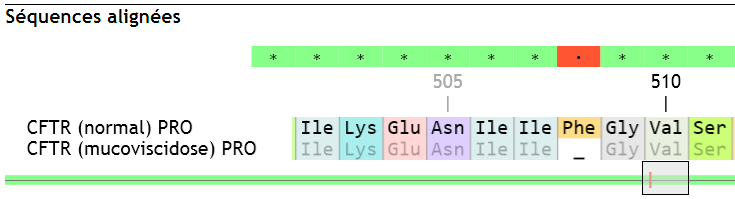
De très nombreuses mutations sont à l’origine de la mucoviscidose mais une est beaucoup plus fréquente que les autres, elle est dite ΔF508.
Pour étudier cette mutation dans Anagène, télécharger le fichier CFTR.edi (clic droit Enregistrer la cible du lien sous...) qui contient les brins non transcrits des parties codantes du gène normal et du gène muté, ou ouvrir directement avec Geniegen2
Résultats d’alignements avec Geniegen2.
La PHE 508 a disparu, il y a eu délétion de TCT (2 dernières bases du 507 et première du 508, ce qui reconstitue un codon ATT pour Isoleucine).
Les séquences alléliques des CFTR sauvage et mutée pour utilisation dans Anagène ou GénieGen sont téléchargeables CFTR.edi (clic droit Enregistrer la cible du lien sous...) ou peuvent être ouvertes en ligne directement avec Geniegen2 : https://www.pedagogie.ac-nice.fr/svt/productions/geniegen2/?load=ADN-HS-CFTR-NORM,ADN-HS-CFTR-MUCO
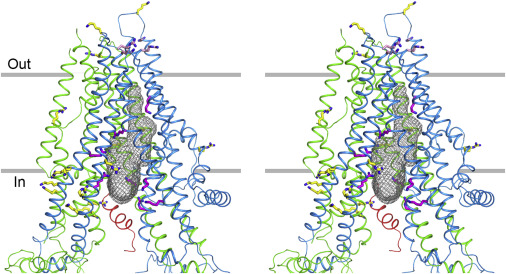
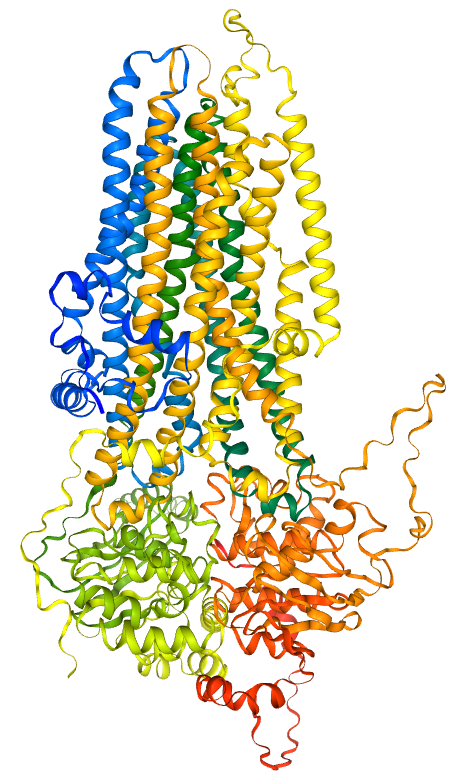
La structure du canal transmembranaire CFTR
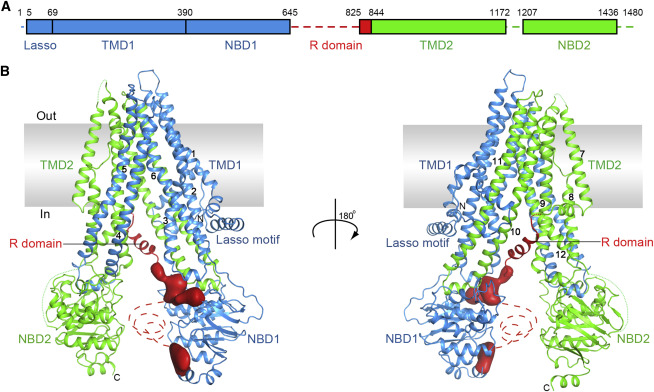
La structure tridimensionnelle du CFTR a été publiée en 2017, en utilisant la technique de cryo-microscopie électronique : Molecular Structure of the Human CFTR Ion Channel, Liu et al., Cell 169 (1), 2017
Structure du CFTR.
TMD = domaine trans-membranaire ; NBD = domaine de liaison au nucléotide ATP ; R = région régulatrice.
Liu et al., Cell 169 (1), 2017
Visualisation de la zone de passage des ions, en grisé.
Liu et al., Cell 169 (1), 2017
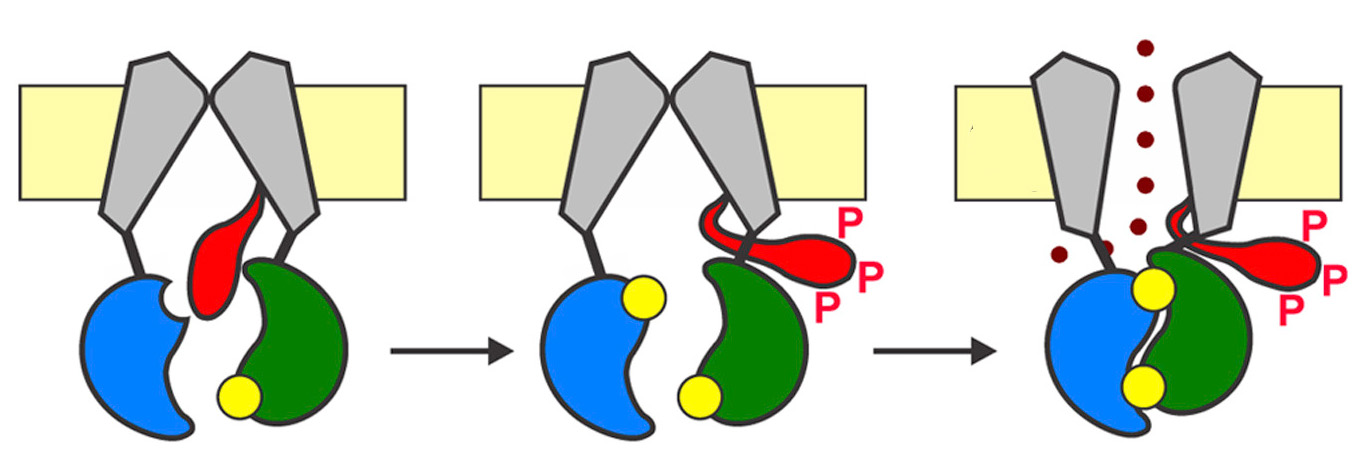
Modèle de fonctionnement montrant les changements de conformation permettant le passage des ions chlorure, après fixation d’ATP sur les domaines NBD et phosphorylation du domaine R.
Points rouges = ions chlorure ; ronds jaune = ATP ; P rouge = groupement phosphate.
Modifié d’après Liu et al., Cell 169 (1), 2017
|
|
|
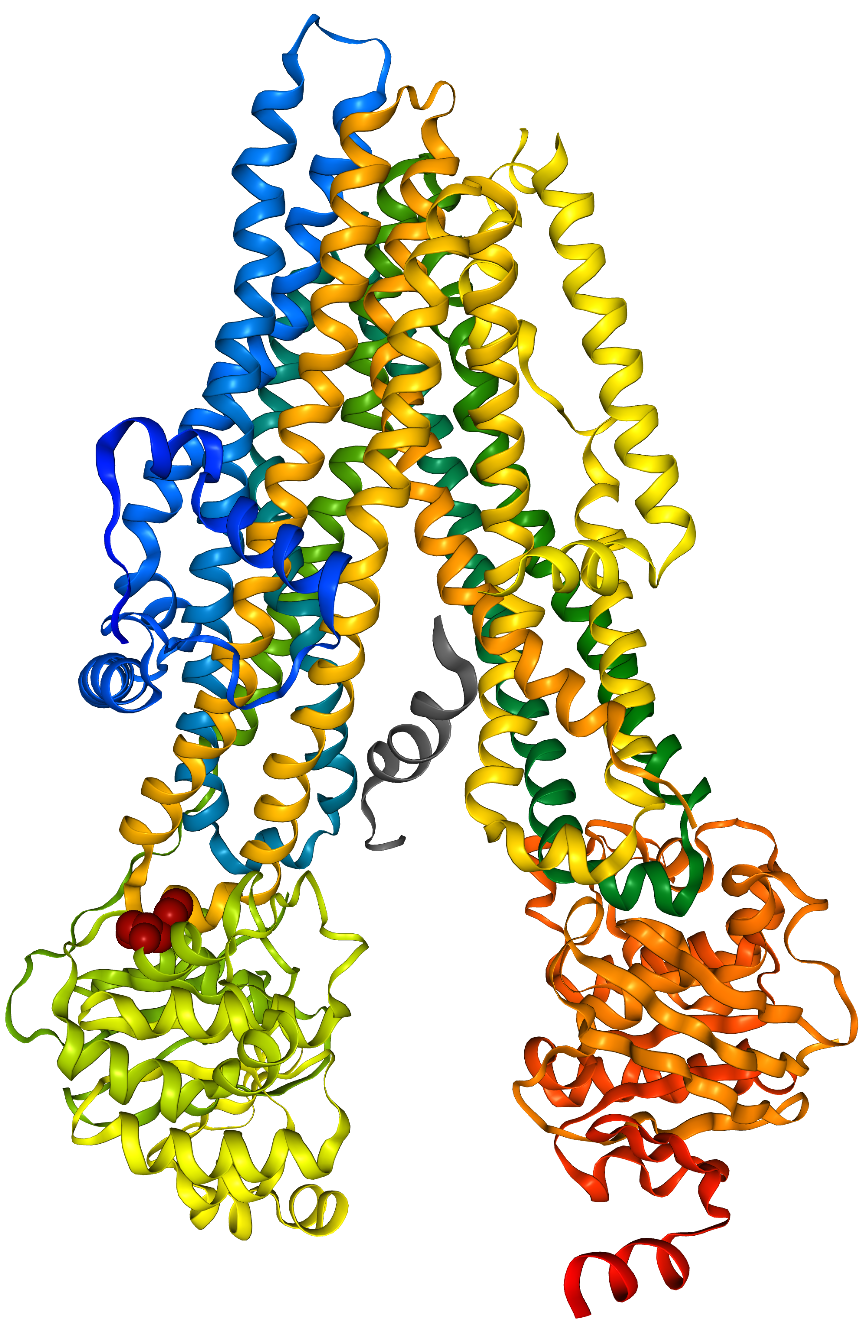
| Modèle de la structure de la protéine CFTR.
Localisation de la Phénylalanine F508, en sphères rouges, à l’interface entre TMD1 et NBD1.
Image obtenue avec Libmol
Le modèle de la structure du CFTR est téléchargeable : CFTR.pdb
|
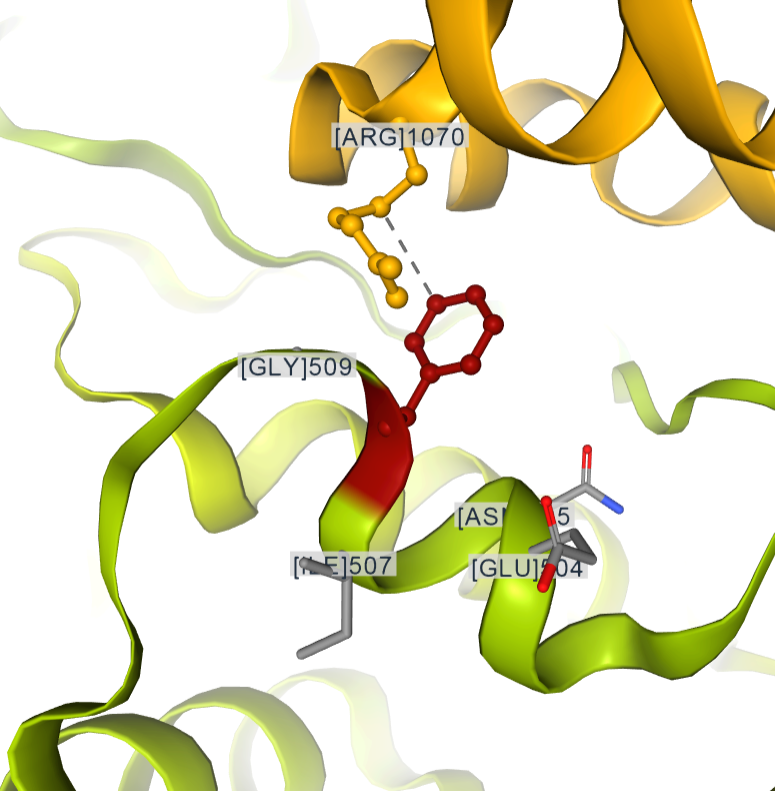
La phénylalanine F508 est figurée en rouge. À cette position elle permettrait d’aider à la conformation de la protéine lors de sa synthèse, et de stabiliser la structure par la suite. L’interaction hydrophobe avec l’Arginine 1070 du domaine TMD1 est mise en évidence. |
| | |
|
|
Modèle de la structure de la protéine CFTR ΔF508.
En absence de la phénylalanine F508, le structure tridimensionnelles du canal est changée.
Image obtenue avec Libmol
Le modèle de la structure du CFTR ΔF508 est téléchargeable : CFTR_deltaF508.pdb
Cette modélisation de la structure du canal CFTR portant la mutation ΔF508 a été réalisée en novembre 2024 avec le serveur AlphaFold 3.
ATTENTION, la structure obtenue est théorique et n’a pas été confirmée par des travaux de recherche.
|
La fréquence des hétérozygotes
Le gène a 2 formes alléliques N (normal dominant) et m (muté récessif), si on nomme p la fréquence dans la population de l’allèle N et q la fréquence dans la population de l’allèle m, on a p + q = 1 puisqu’il n’y a que 2 allèles.
Si les unions entre les individus se font au hasard, il y aura un mélange aléatoire de gamètes porteurs de l’allèle N ou de l’allèle m, conduisant à 3 génotypes possibles : NN, Nm ou mm.
Grâce à un échiquier de croisement représentant des rencontres aléatoires de gamètes normaux et mutés, on peut trouver les fréquences des homozygotes et des hétérozygotes.
| |
N (p) |
m (q) |
| N (p) |
N//N (p2) |
N//m (pq) |
| m (q) |
N//m (pq) |
m//m (q2) |
Comme la fréquence de la mucoviscidose est en moyenne de 1/2500 quelle est la fréquence des hétérozygotes ?
La fréquence de 1/2500 correspond à q2 donc q = 1/50, p est donc 1 - 1/50 soit environ 1. La fréquence des hétérozygotes est 2pq soit 2 x 1 x 1/50 = 1/25.
Une personne sur 25 est porteuse de l’allèle muté. Cette fréquence élevée conduit à rechercher des explications.
Les liens entre mucoviscidose et choléra
Le choléra est dû à une bactérie (Vibrio cholerae) qui colonise l’intestin et provoque une diarrhée plus ou moins sévère pouvant entraîner la mort.
Il existe des différences importantes de sensibilité au choléra entre les populations exposées à la bactérie. Près de 90 % des sujets infectés n’ont pas de symptômes et éliminent les bactéries par voie intestinale, 10 % auront une diarrhée dont environ 1 % présenteront un choléra sévère.
Les porteurs sains, qui semblent jouer un rôle majeur dans la dissémination de l’épidémie, témoignent d’une sensibilité génétique variable des sujets exposés.
La bactérie se colle contre la membrane des cellules intestinales et sécrète localement une toxine qui pénètre dans ces cellules ce qui déclenche des réactions biochimiques perturbant le fonctionnement de la molécule CFTR. Ce canal laisse alors passer des quantités plus importantes d’ions Cl- vers la lumière intestinale. Les ions Cl- sont accompagnés par de nombreuses molécules d’eau qui provoquent une diarrhée.
Les liens entre mucoviscidose et typhoïde
Salmonella typhi est la bactérie responsable de la typhoïde (inflammation grave du tube digestif). Pour que cette maladie se déclare il est nécessaire que la bactérie pénètre dans les cellules intestinales.
Certaines expériences auraient été réalisées sur des souris transgéniques chez qui on a intégré le gène humain sous la forme normale ou sous la forme mutée ΔF508.
Si on fait absorber par voie buccale des bactéries Salmonella typhi à ces souris, les résultats peuvent être résumés comme suit :
| Souris homozygote pour l'allèle muté |
0% de cellules infectées |
| Souris homozygote pour l'allèle normal |
100% de cellules infectées |
| Souris hétérozygote pour le gène étudié |
14% de cellules infectées |
Le test de la sueur
Il consiste à mesurer le taux de chlorures dans la sueur. Les résultats sont interprétés de la manière suivante :
| Taux de chlorures |
Résultat du test |
| inférieur à 40 mmol.L-1 |
négatif |
| compris entre 40 mmol.L-1 et 60 mmol.L-1 |
douteux |
| supérieur à 60 mmol.L-1 |
positif |
Les individus malades ont donc une sueur plus riche en chlorures que les individus sains.
On peut rechercher la cohérence de ce test avec les informations obtenues dans les pages précédentes sachant que le canal CFTR des glandes sudoripares sert à la réabsorption des ions Cl-.